

Beta cell transcription-splicing axis is disrupted in type 2 diabetes
Mutations in a single gene, HNF1A, are known to cause MODY3, a rare, early onset form of diabetes. Smaller scale mutations in the very same gene are also common and quietly nudge millions of people toward type-2 diabetes. A study published in Cell Metabolism reveals why.
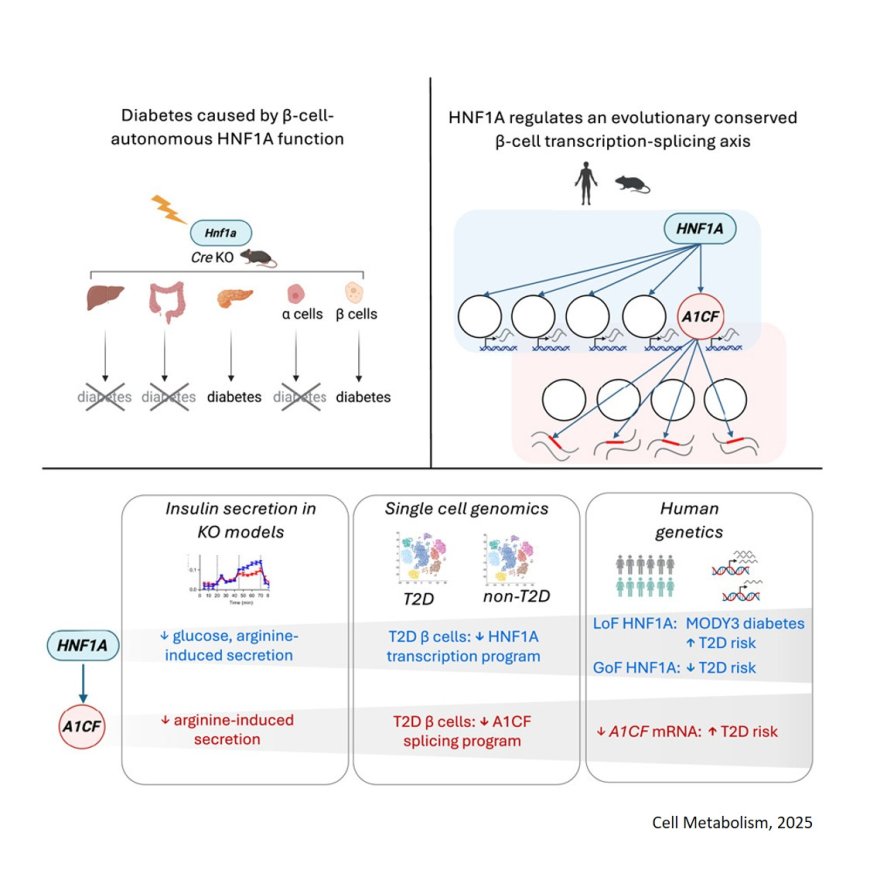
Researchers show it’s fundamentally a problem of insulin-producing β‑cells. Using mouse models, they switched HNF1A off in different tissues and cell types including the liver, the gut and both α and β‑cells in the pancreas, one at a time. Blood glucose levels were only affected when the gene was deleted in β‑cells.
HNF1A is a known transcription factor, meaning its job is to bind to DNA and finetune the expression of other genes. The study found that deleting HNF1A in either human or mouse β‑cells affected the expression of more than one hundred genes, many of which encode for the molecular parts required to transport and release insulin.
The team also found that one of the direct targets of HNF1A happens to be A1CF, a second gene which assembles (or splices) RNA molecules before they’re turned into proteins. When HNF1A is mutated, A1CF levels collapse and the β‑cell’s RNA molecules are scrambled on a massive scale, accumulating between 1,900 and 2,300 different RNA splicing mistakes.
“When HNF1A fails, two things go wrong at once. Hundreds of genes that depend on it begin to work incorrectly. That alone is enough to weaken insulin secretion, but the loss of A1CF means that the RNAs that are still made now get spliced incorrectly. Both layers matter, but the first hit is broader and sets the stage while the second piles on extra dysfunction,” says a co-first author of the study.
Studying human pancreatic cells painted a similar picture. In healthy donors, a robust population of β‑cells buzzed with HNF1A and A1CF activity, but in donors with type-2 diabetes, researchers observed a major increase in populations of cells with low HNF1A and A1CF activity.
“In people with type-2 diabetes, for every high-functioning β‑cell we found about eight low-functioning ones, while healthy donors had a healthier ratio of one to one. It’s a dramatic shift that shows how a single mutation can cascade into the loss of function of entire tissues and organs,” says another co-first author of the study.
The discoveries made by the study offer a new druggable foothold for diabetes, both for MODY3, which affects around 0.03% of the general population, and type-2 diabetes, which has become so widespread that more than one in nine adults, around 600 million people worldwide, now live with the disease.
Diseases like spinal muscular dystrophy have become treatable by fixing scrambled RNA messages. Because the diabetes defect uncovered here is an RNA splicing problem, the same strategy could, in principle, be used to “re‑edit” β‑cell RNA molecules, tackling one of the root causes of the disease.
“Existing therapies for diabetes try to lower blood sugar with different strategies without correcting underlying defects. The RNA defects we found are patchable, offering a rare, clear target for an incredibly complex disease,” explains the corresponding author of the study.
However, type-2 diabetes is driven by many genes and lifestyle factors. “We can now say this defective program has a causal contribution,” says the author, “but there are other molecular defects that also need to be addressed. This is only one piece of a larger puzzle that we’ll also have to solve.”
The research group next plans to build what he calls a molecular parts list of the genetic chain of command, hoping to flag every possible protein and RNA molecule that could serve as a potential drug target. “The goal is to pinpoint the most practical targets for new β‑cell therapies, so we can translate these insights into effective treatments,” concludes the author.
https://www.cell.com/cell-metabolism/fulltext/S1550-4131(25)00334-1
https://sciencemission.com/beta-cell-transcription-splicing-axis