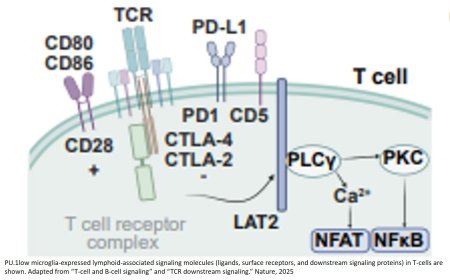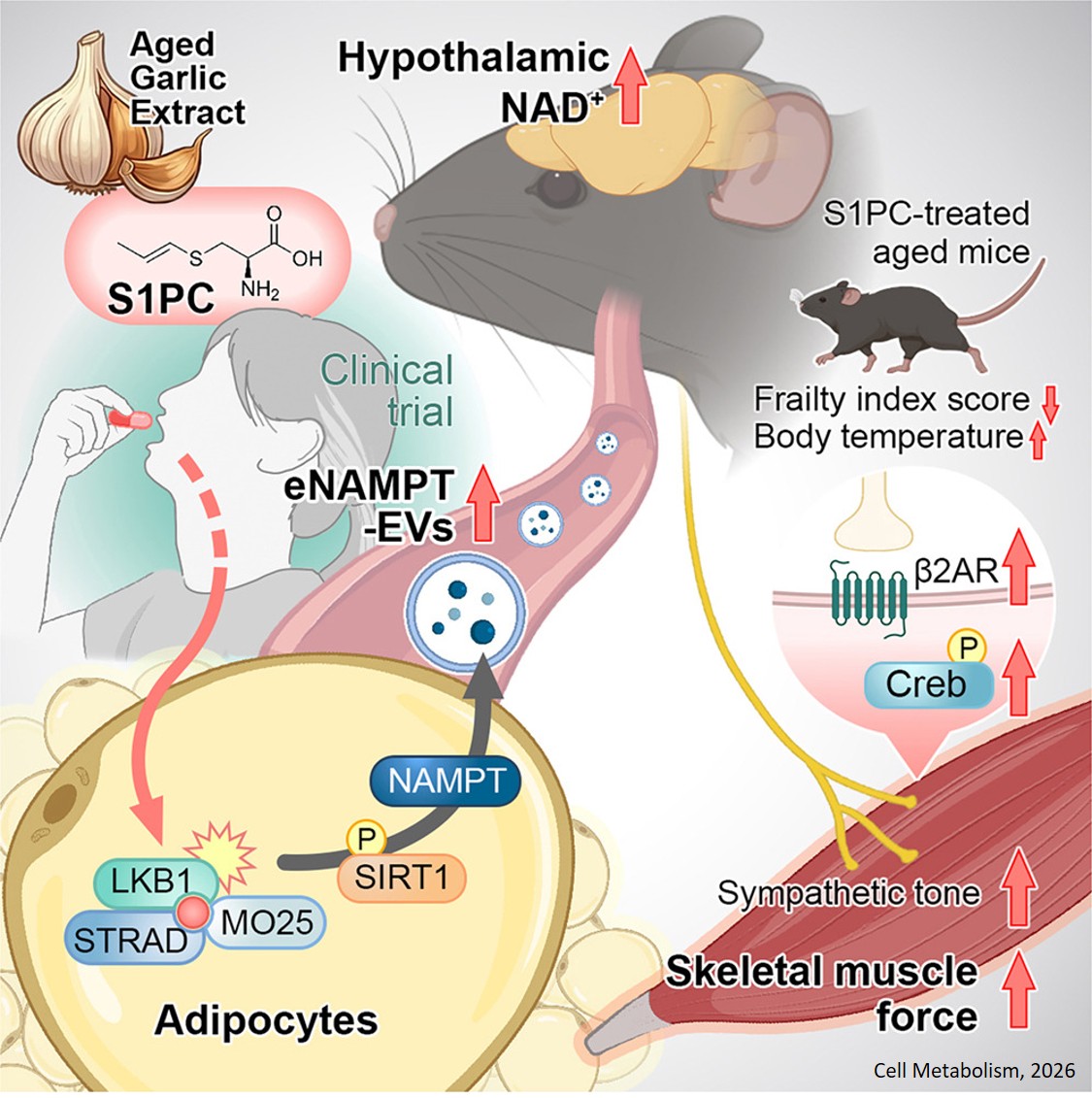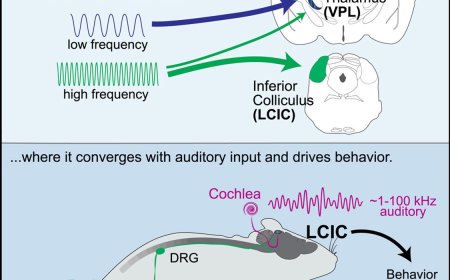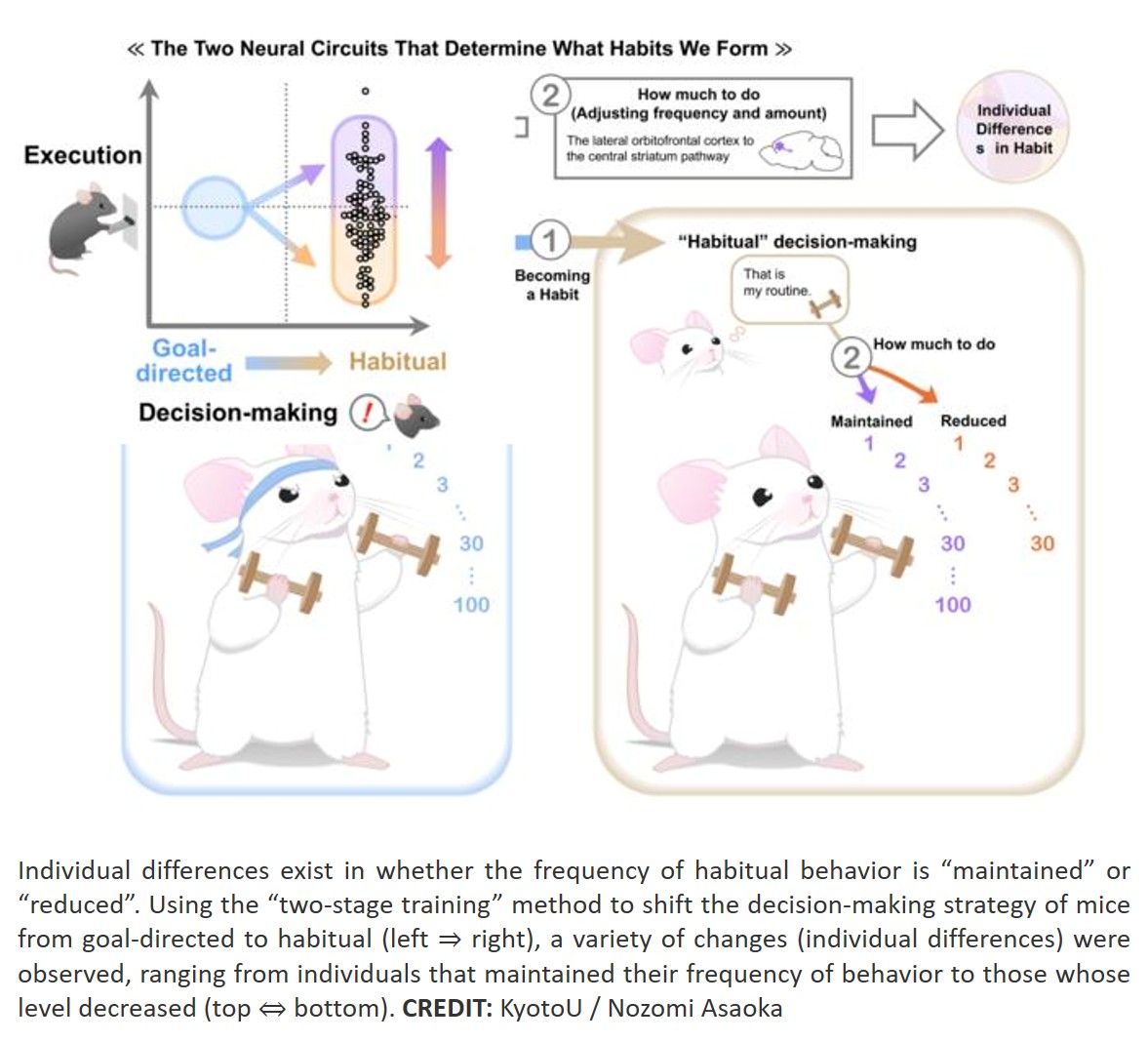
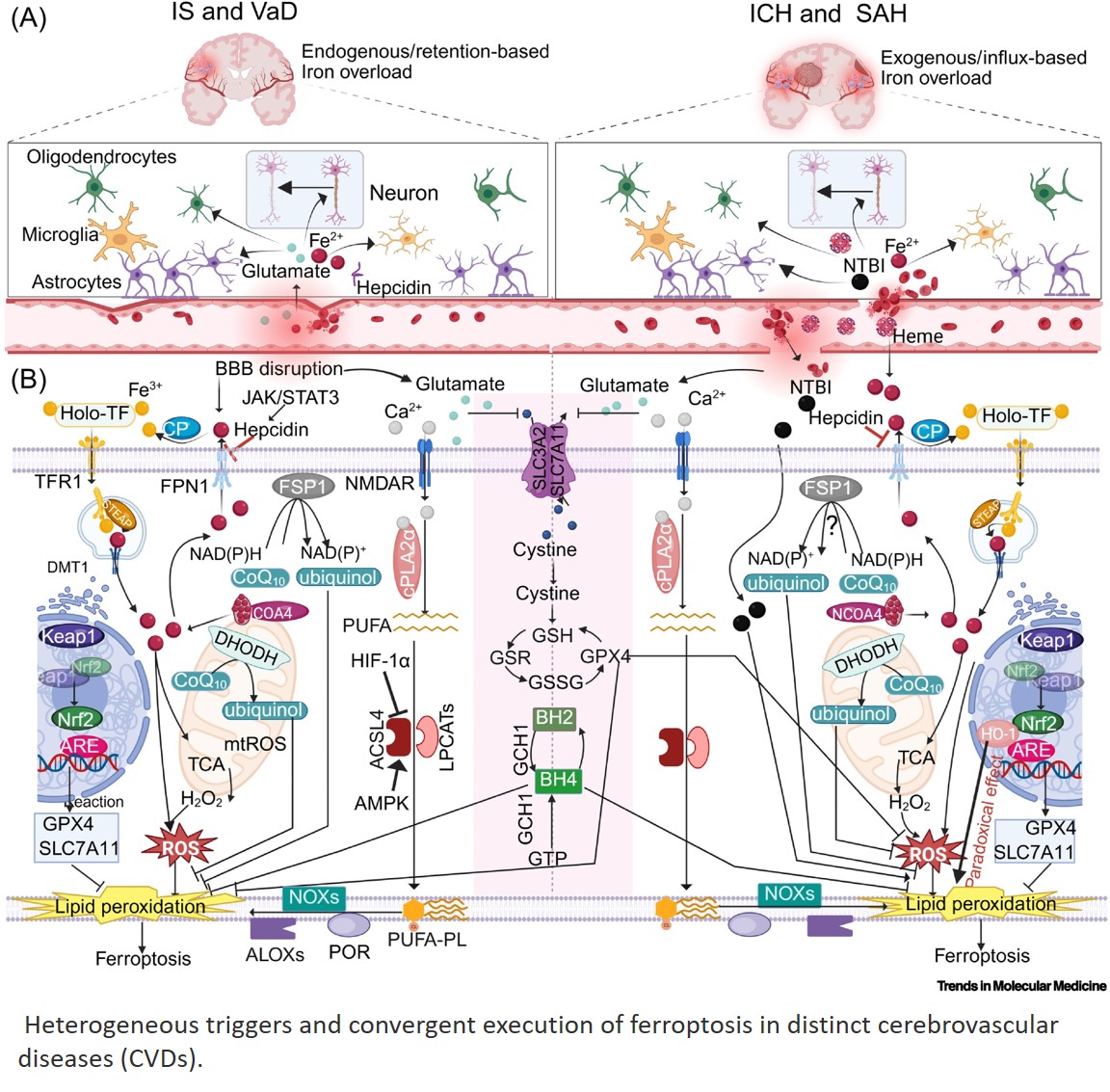
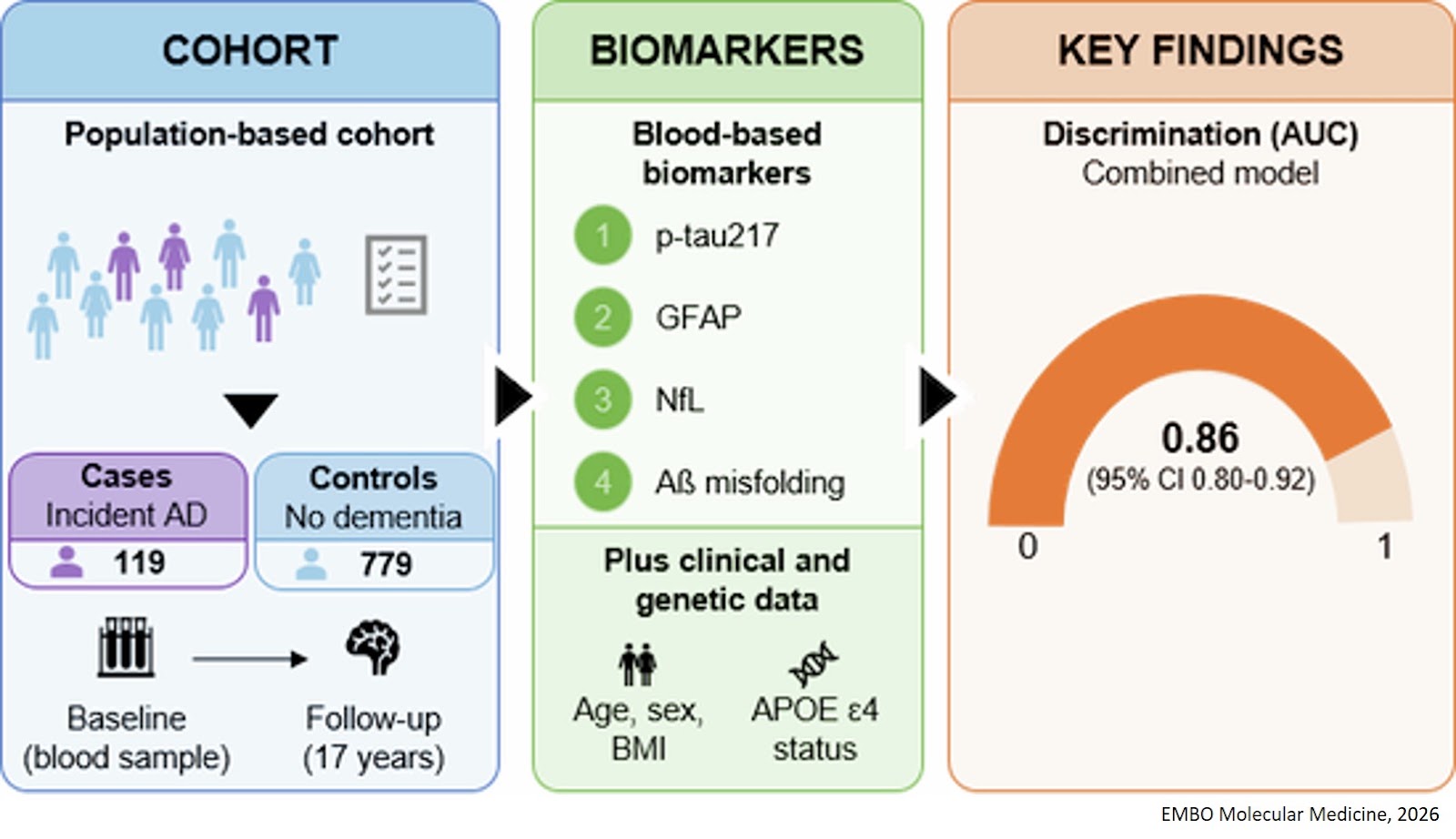
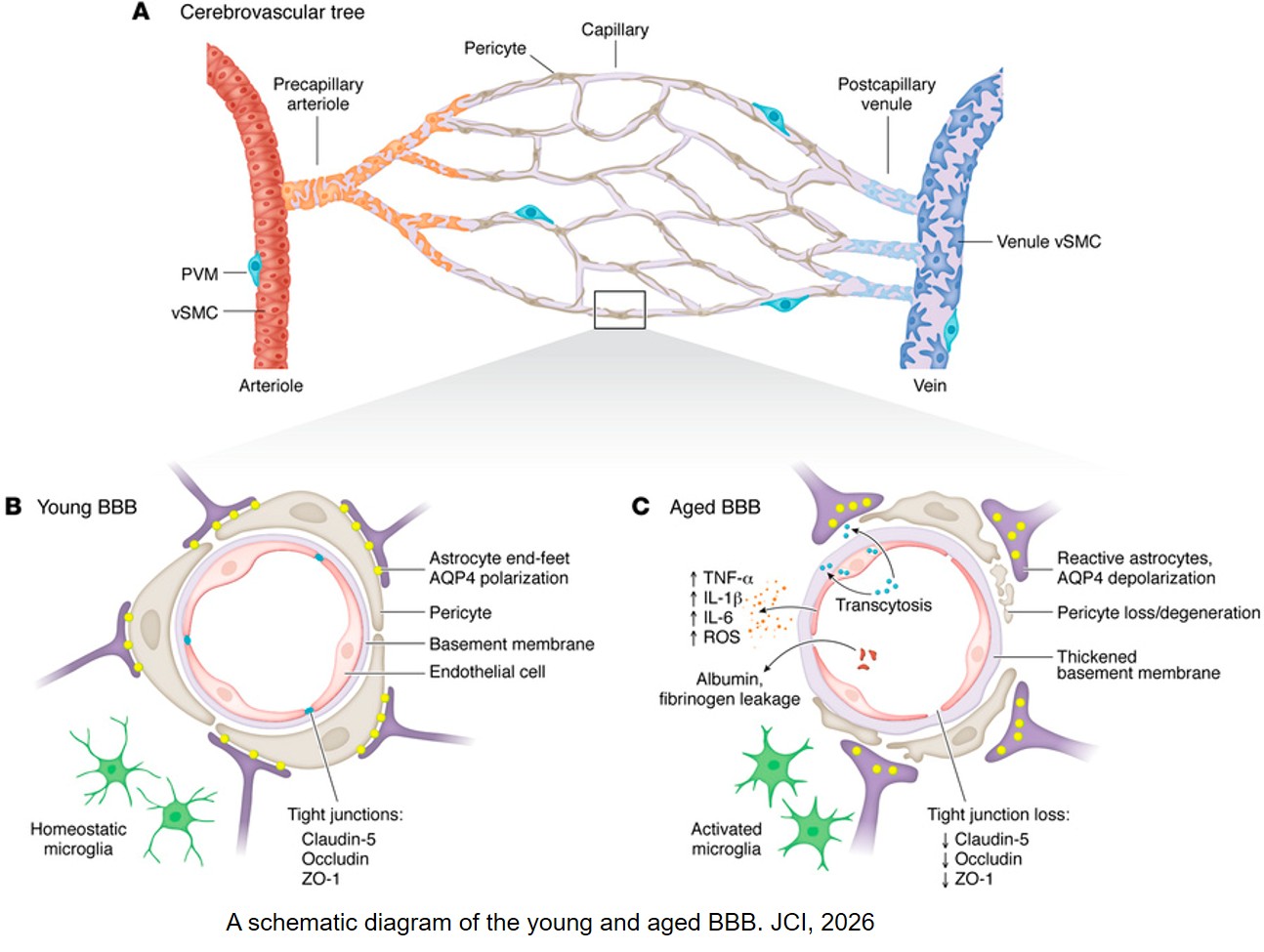
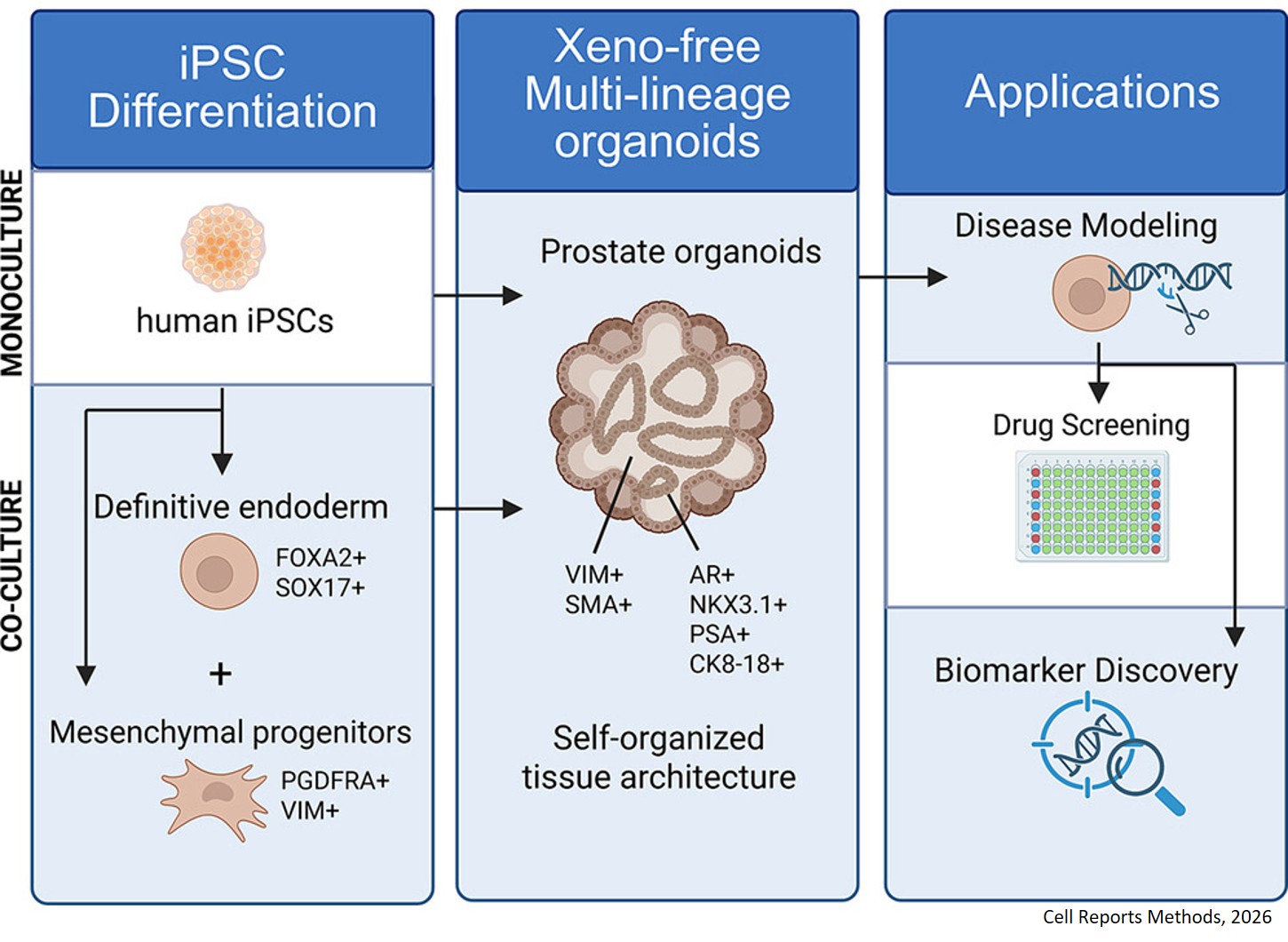


Histone modifications and dysregulation of transcriptome prior to clinical symptoms in Rett syndrome
Scientists investigating the severe developmental disorder known as Rett syndrome have discovered a series of crucial molecular changes that occur long before symptoms appear. The findings could be used to develop better treatments for the devastating, life-shortening condition, the researchers say.
Rett syndrome strikes girls almost exclusively. Children with Rett initially appear healthy and appear to develop normally for the first six to 18 months before beginning to regress and lose previously acquired skills. For example, children who crawl can become unable to do so, and language skills decline. Other symptoms of Rett include difficulty eating, seizures, “floppy” limbs and the repetitive hand movements that are the disease’s hallmark. These symptoms can range from mild to severe. Life expectancy varies, but many people with Rett die by their 40s or 50s.
The researchers began investigating how mutations in a particular gene, MECP2, trigger the development of Rett.
That investigation has revealed a whole “cascade” of molecular changes that fundamentally alter how genes work in brain cells. In particular, the scientists discovered that the cascade causes far-reaching, “circuit-level” problems in the hippocampus, an area of the brain vital for memory and learning. These sweeping changes cause brain cells called neurons to begin malfunctioning, the authors determined.
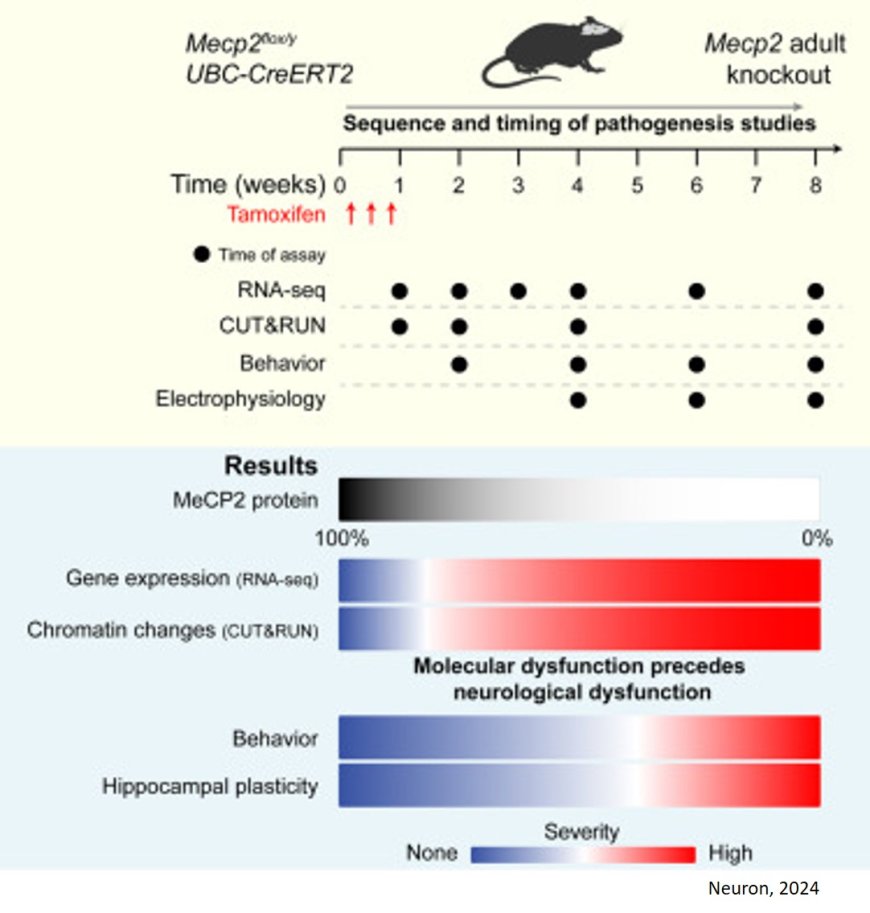
The researchers found that loss of MeCP2 causes immediate and bidirectional progressive dysregulation of the transcriptome. To understand what drives gene downregulation, they profiled genome-wide histone modifications and found that a decrease in histone H3 acetylation (ac) at downregulated genes is among the earliest molecular changes occurring well before any measurable deficiencies in electrophysiology and neurological function.
“We artificially triggered the onset of Rett syndrome symptoms in mice to precisely map the sequence of events that occurs when MECP2 is malfunctioning. Our study uncovered a core set of genes that are disrupted very early on before any overt symptoms have presented,” said the author. “These genes might be drivers of Rett syndrome symptoms downstream of MECP2 whose expression levels could be important for normal brain function as well.”
The discovery of these molecular changes – and the specific mechanisms responsible for the changes – sheds much-needed light on the development of Rett syndrome. It also sets the stage for new and better ways to treat the condition. For example, there is great excitement about the potential of gene therapy to restore the MECP2 gene’s function in children with Rett. The challenge, however, is that augmenting the gene’s activity too much would prove toxic to brain cells.
Doctors need ways to monitor the activity of the gene, and this research could ultimately provide that. For example, doctors might be able to monitor biological markers, or “biomarkers,” the scientists have identified that reflect whether the MECP2 gene is functioning at an appropriate level.
While much more research needs to be done before the findings could be translated into treatments, the authors are excited about the potential his findings hold.
“We discovered several candidate biomarkers sensitive to MECP2 levels that could be the key to developing safe gene therapies for Rett,” the author said. “Our study more broadly demonstrates the importance of cataloging and understanding the earliest biological events that occur during symptom onset in neurodevelopmental disorders.”