
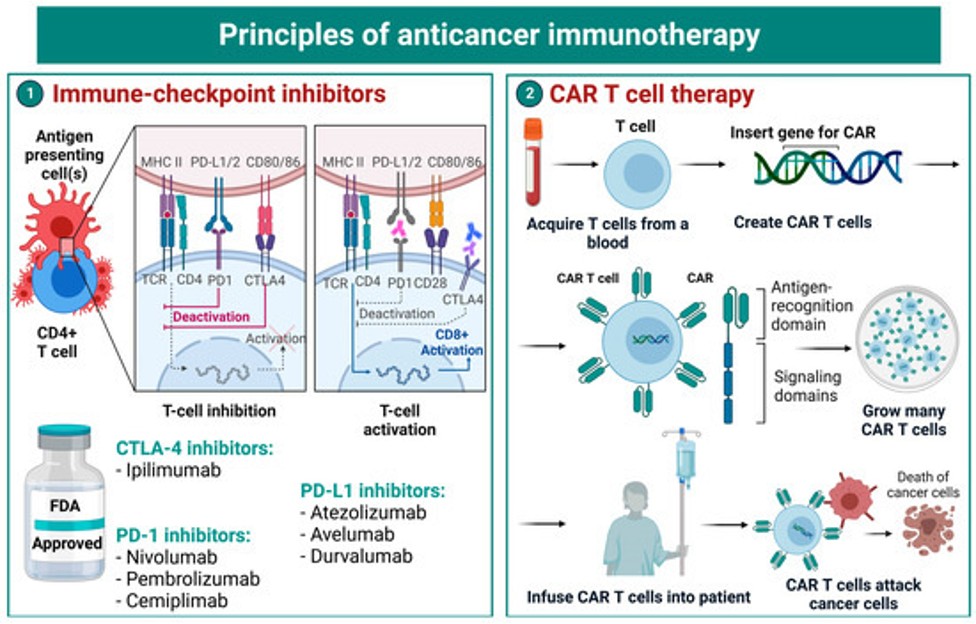

Mitochondrial fission and traumatic brain injury
Violent blows or jolts to the head can cause traumatic brain injury (TBI), and there are currently about five million people in the U.S. living with chronic neurodegeneration and related impairments due to TBI. In addition to cognitive and mental health impairment, chronic neurodegeneration may also contribute to why TBI increases the risk of age-related neurodegenerative diseases, such as Alzheimer's or Parkinson's disease. It could also play a role in chronic traumatic encephalopathy (CTE).
Due to the lack of understanding of why acute TBI transitions into chronic neurodegeneration, however, there are currently no treatments that protect patients from this outcome. Now, researchers have moved a step closer to finding answers in a study recently published in Cell Reports Medicine.
“We started with the hypothesis that TBI might pathologically impair the balance of mitochondrial fission and fusion,” explained the lead author of the study. “The normal homeostatic balance of mitochondrial fission and fusion is how mitochondria consistently produce enough energy for the cell while also sequestering and disposing damaged parts. Given the very high energy demands of the brain, this is particularly important for brain health across our lifespan.”
The process is governed by the interaction of two cellular proteins: Fis1 and Drp1. It was previously shown that other neurodegenerative diseases, including Alzheimer's disease (AD) and Huntington's disease, display pathologically elevated mitochondrial fission due to elevated expression of Drp1. Here, the research team discovered that mitochondrial fission is pathologically elevated in mouse and human TBI as well, but that it is caused by increased expression of Fis1, rather than Drp1.
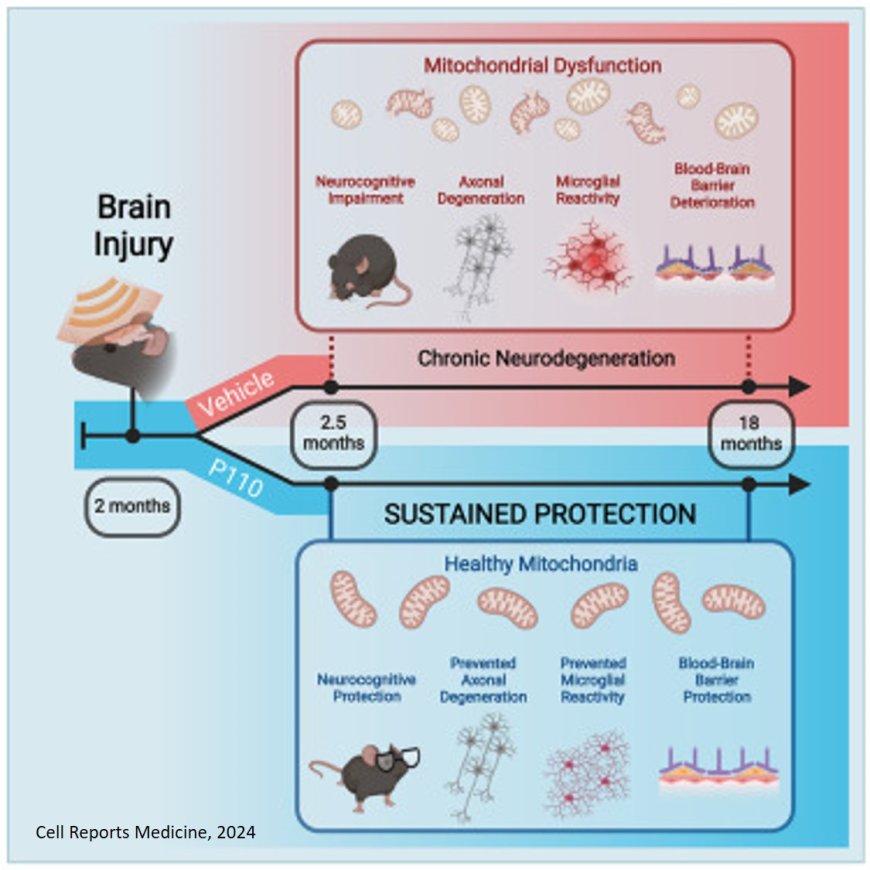
They next tested whether pharmacologically reducing excessive mitochondrial fission for only two weeks after TBI, by administering a small peptide agent named P110 that blocks the interaction of Fis1 and Drp1, might halt this process and protect the brain. P110 was previously discovered and developed by a co-senior author.
“Brief P110 treatment during the acute time period after TBI permanently normalized mitochondrial fission / fusion and prevented subsequent harm to the brain, including oxidative damage, blood-brain barrier deterioration, axonal degeneration, and cognitive impairment, 17 months later. This is equivalent to many decades in people,” explained the senior author of the study. “The same treatment administered much later, however, had no protective effect. Thus, there is a critical time window after TBI wherein this treatment can be effective.”
The team hopes that P110 or a related compound will be tested clinically in acute TBI patients. “Next steps in the basic science research, on the other hand, involve further utilization of this model to yield additional new insights into understanding the pathophysiology and treatment opportunities for this important problem,” explained the author.
In addition to extending their investigation to additional different preclinical models of TBI, the research team also plans to investigate whether the mechanism they discovered could play a role in why TBI accelerates AD. They speculate that the combination of increasing two components of the same system (increased Fis1 in TBI and increased Drp1 in AD) could cause a synergistic deleterious effect that significantly advances the development and severity of AD after patients have experienced a TBI.
https://www.cell.com/cell-reports-medicine/fulltext/S2666-3791(24)00436-1