

What changes happen in the aging brain?
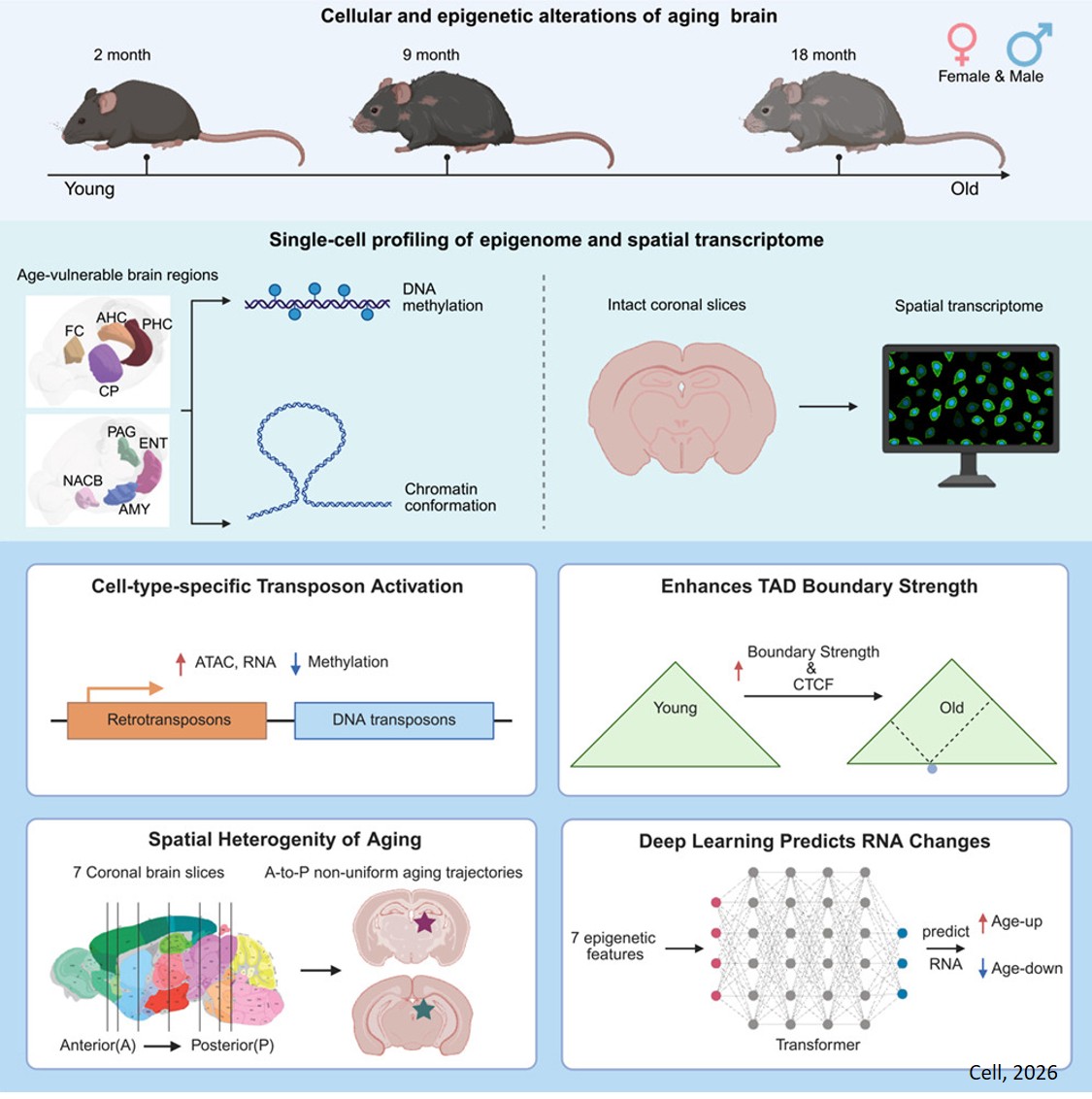
One major mechanistic influence on aging is epigenetic change: the way small chemical tags on top of our base genetic code shift over time to alter gene expression. The researchers created the most comprehensive single-cell atlas to date of epigenetic changes in the aging mouse brain, revealing how DNA methylation, genome structure, and gene activity change across brain regions and cell types. The new atlas represents eight brain regions and 36 distinct brain cell types, with over 200,000 single cells profiled across methylation and chromatin conformation assays, plus nearly 900,000 cells captured with spatial transcriptomics.
The atlas’ contents have already revealed clear epigenetic differences between different age groups, as well as allowed the researchers to develop novel deep-learning models that predict age-related gene expression changes. Published in Cell, the atlas is now publicly available on Amazon Web Services (AWS) and Gene Expression Omnibus (GEO), where it will serve as a critical reference framework for interpreting human brain datasets, including those generated by the National Institute of Health’s Brain Research Through Advancing Innovative Neurotechnologies (BRAIN) Initiative.
“Age-related brain changes, particularly in regions critical for attention, memory, emotion, and motor functions, severely impact life quality,” says co-corresponding author. “By mapping how the epigenome changes across individual brain cell types as animals age, we now have a framework for understanding how aging reshapes the brain at the molecular level. This resource should help researchers pinpoint mechanisms that contribute to neurodegenerative disease.”
With age comes four molecular hallmarks: chronic inflammation, mitochondrial dysfunction, genome instability, and epigenetic changes. Recent findings have pinpointed the epigenome as a major driver of physiological aging. And one type of epigenetic change called methylation has been associated with neuronal function, behavior, and disease. If scientists could connect the dots between methylation changes and adverse age-related outcomes, they could begin to engineer solutions that reverse those changes and rescue health.
But generating useful methylation data isn’t as simple as sampling a few brain cells and drawing generalized conclusions. The brain has many different regions and cell types, which each must be considered to get a full picture of what’s going on.
"The brain is so interconnected, with different regions controlling different functions and aging at different speeds at the cell type level,” says the co-corresponding author. “We can see how interconnected the brain is in conditions like Parkinson’s, where the death of one group of neurons spirals into an entire circuit malfunctioning and then the tremors and cognitive effects we see in patients. So, the importance of having a cell type-specific understanding of aging will bring more granular knowledge that will expand therapeutic possibilities.”
Bulk analysis of brain cells loses cell type specificity, making single-cell analyses a powerful tool. So, the researchers set out to create the most comprehensive single-cell, multi-omic brain imaging data set to date. On top of methylation, they surveyed another genome-regulating mechanism called chromatin conformation—the 3D shape of the genome. They also used cutting-edge spatial transcriptomics technology to map gene expression while preserving spatial context within the sampled brain tissue.
“What makes this work innovative is, above all, its spatial dimension,” explains the first author. “Spatial resolution reveals which regions and local microenvironments are most vulnerable to aging, how cell-type composition shifts across brain areas over time, and how neighboring cells may influence one another's aging trajectories. The scale of the spatial dataset—nearly 900,000 spatial transcriptome cells—is itself unprecedented for a longitudinal aging study.”
Using a mouse model of aging, the team collected methylation data on 132,551 single brain cells and joint methylation-chromatin conformation data on 72,666 brain cells. Together, 36 major cell types were represented. This dataset was published in full on AWS and GEO in December 2025. Hosting this massive dataset of nearly 900,000 spatially resolved cells on AWS ensures both accessibility and immediacy. Typically, such massive amounts of data require significant computational power to access, but cloud hosting tears down those infrastructure barriers.
“The AWS Open Data program covers storage costs and places this dataset alongside other major neuroscience resources like the Allen Brain Atlas and the Seattle Alzheimer's Disease Brain Cell Atlas, making it part of an interconnected ecosystem of publicly accessible brain data,” adds the author. “Researchers in aging, neurodegeneration, and spatial genomics can build on this resource immediately, accelerating the pace of discovery well beyond what a single lab could achieve.”
First, the methylation data revealed that age-related methylation changes were more pronounced in non-neuronal cells. The team found that transposable elements—sometimes called ‘jumping genes’—lose DNA methylation as cells age, suggesting that normally silenced genomic elements become more active in the aging brain. Jumping genes are repetitive DNA sequences that make up around half of the human genome, and their expression can lead to dysfunction and age-related decline. This finding is consistent with the idea that epigenetic changes may contribute to aging-related cellular dysfunction.
The chromatin confirmation data revealed further changes during aging. Notably, the researchers were able to identify a new biomarker for brain aging: increased strength at topologically associating domain (TAD) boundaries and greater accessibility at related CTCF binding sites. The massive amount of information contained in a genome is organized using TADs, which are simply smaller stretches of DNA that work together. CTCF is a protein that binds to boundaries on either end of TADs, assisting in their organization.
Then came the time to fold in the spatial transcriptomics insights. Nearly 900,000 cells were used to trace differences between aging in different brain regions and cell types.
“The same cell type ages differently depending on its location; for instance, non-neuronal cells in the back of the brain show more inflammation than those in the front parts,” says the author. “This data really underscores the variability in aging even among the same cell type, emphasizing the importance of cell and brain region-level specificity in unraveling the complexities of aging.”
The team has already gleaned impressive insight from the dataset. For example, they developed deep learning methods to predict gene expression using future multi-omic epigenetic data, thereby laying the groundwork for the future development of a virtual brain aging model. And more exciting insights are on the way.
The atlas is available online now for anyone to use. By making these sorts of resources accessible to all, the scientists hope to see their findings accelerated by the power of global collaboration.
https://www.cell.com/cell/fulltext/S0092-8674(26)00222-9
https://sciencemission.com/aging-mouse-brain