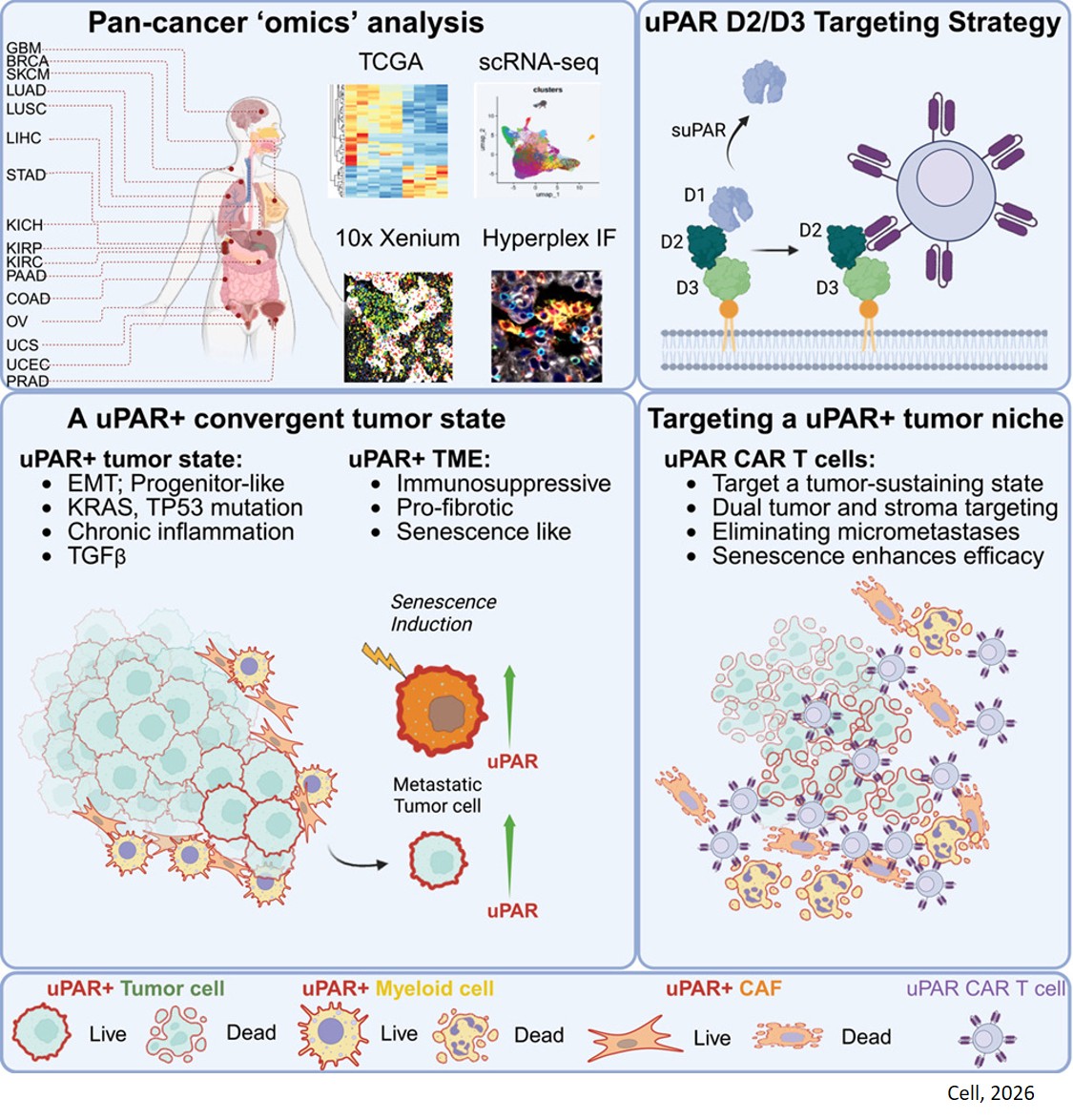


Flipping the switch from benign growth to pancreatic cancer
As we age, our cells accumulate genetic changes — mutations — some of which open the door to cancer. Scientists call these mutations “oncogenic,” meaning “tumor producing.”
By our senior years, we each may harbor as many as 100 billion cells with at least one oncogenic mutation, researchers have estimated. But many people never develop cancer — and scientists have long worked to understand why only a tiny fraction of these mutated cells ever progress into tumors.
Now, a study reveals new details about how pancreatic cells carrying oncogenic mutations transition from benign to malignant.
“It’s increasingly clear that the progression of a malignant tumor isn’t just about transformations happening within cells, but about coordinated interactions between cells and nearby tissues that develop ecosystems to support tumors,” says study co-senior author.
Pancreatic ductal adenocarcinoma is one of the deadliest cancers, with a five-year survival rate hovering around 13%. It also tends to develop through recognizable stages — from early benign growths to invasive cancer — making it a useful model for studying how cells transition from pre-cancer to cancer.
In pancreatic cancer, one genetic mutation stands out. The KRAS gene is altered in nearly all pancreatic ductal adenocarcinomas, the disease’s most common and deadly form.
When cells in the pancreas are injured by inflammation caused by, for example, pancreatitis, they normally enter a temporary repair program and then return to normal. But when the cells contain an oncogenic KRAS mutation, that repair program can get stuck in the “on” position, keeping cells in an unusually flexible state, instead of resolving normally.
Still, these precancerous cells with KRAS mutations can remain benign for a long time.
To find out exactly what is required for them to become malignant, the research team used a variety of advanced technologies.
“Essentially, what we saw was a tug-of-war between oncogenes and tumor suppressor genes,” says the study’s lead author.
The team tracked pancreatic cells with KRAS mutations and used a marker that let them spot cells that had also lost the tumor suppressor p53. The p53 gene is known as the “guardian of the genome” because it turns on protective programs when cells show signs of DNA damage. Therefore, it was important for researchers to compare tumor cells to precancerous cells both with and without working p53.
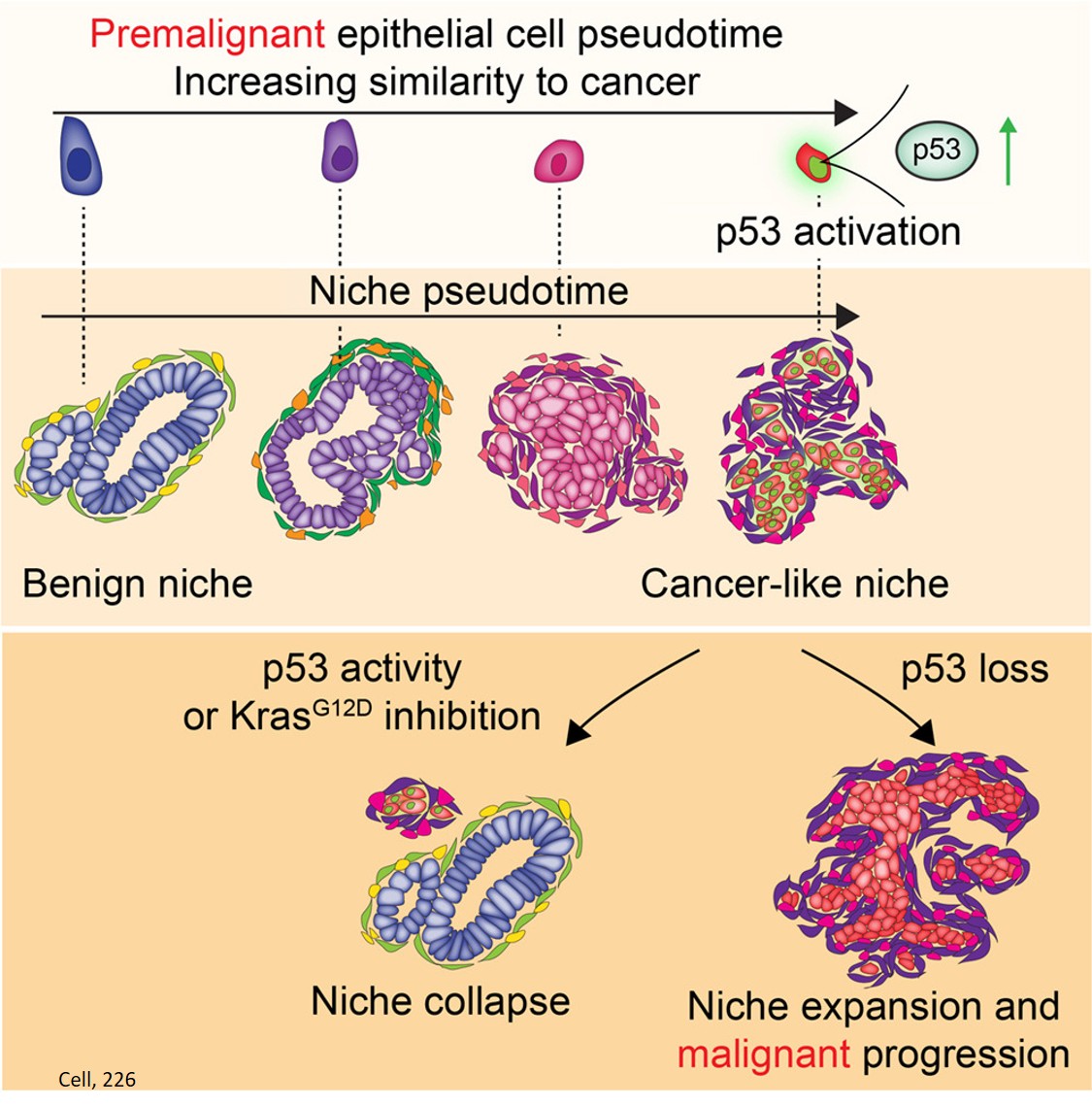
Using these techniques, the scientists were able to identify a subset of precancerous cells that most closely resembled tumor cells — cells right at the tipping point of becoming malignant.
Counterintuitively, they found that these cells also had the most strongly activated tumor-suppressors: p53 and two others — CDKN2A and SMAD4.
This combination of opposing forces — a high oncogenic drive plus a high tumor-suppressor response — often results in a stalemate, putting cells into what scientists call “senescence,” a kind of emergency brake that prevents cells from continuing to divide when growth signals are abnormally strong.
“The body is seeing this particular cell state as dangerous — and putting extra pressure on it to stop,” the author says.
But if the body can’t get cells unstuck from this state, the cells begin to reshape their surroundings and set the stage for cancer to emerge — especially when additional genetic changes knock out p53.
“What we’re finding is that p53 is even more than the guardian of the genome, it’s the guardian of plasticity,” says another author. “Cells need to become plastic for regeneration and repair to happen, but p53 is there to pump the brakes if they try to go rogue. Without p53, this flexibility can be hijacked by cancer.”
Pancreatic tumors are among the most difficult to treat because they’re protected by a dense weave of scarlike tissue and support cells.
The study shows that pre-cancers start to make these protective neighborhoods surprisingly early — well before a full-blown tumor appears.
The team was able to see how precancerous lesions and their surroundings change together using two techniques. The first is spatial transcriptomics, which allows researchers to visualize where specific cells and genes are active within a tissue. They also developed a new computational approach to modeling changes in the local neighborhood around a given cell.
“For the first time, this new approach allowed us to reconstruct dynamic changes in a cellular neighborhood — and allowed us to see how this rare subset of cells makes changes in nearby cells that support the development of a tumor,” the author says.
As researchers further investigated the cellular neighborhood, they discovered cells stuck in the injury repair state increasingly engage two key players, which undergo coordinated changes:
· Fibroblasts, which begin to build up fibrous, scarlike tissue.
· And myeloid immune cells, which start sending “stand down” signals that keep other immune cells from attacking.
“The neighborhoods around these pre-cancers start to look very much like the environment you see in invasive pancreatic cancer,” the author says. “This repair program allows the pre-cancer to communicate with its neighbors to reshape its local environment in ways that pave the way for cancer to take hold.”
The new study that showed how cell-to-cell communication expands during the early development of pancreatic cancer.
One of the reasons pancreatic cancer has such a low survival rate is that it’s usually caught late, when the tumors are well-established and have started to spread.
The encouraging news is that this early ecosystem might present an opportunity for medical intervention, the researchers say.
The scientists gave mice a short treatment with a KRAS-blocking drug, which not only eliminated the premalignant cells, it disrupted the niche around them, too.
“A three-day treatment in mice paused cancer development for months, which would be the equivalent of years on a human timescale,” the author says.
Not only do p53 mutations happen in about half of all cancers, those cancers are also among the most aggressive and often there are no good treatment options.
“So, it’s been a major goal of the field to develop strategies against these cancers,” the author says.
Many cancer drugs work by shutting down oncogenes that are overactive, but trying to compensate for p53’s loss has presented a much tougher clinical challenge.
“Some efforts have tried to mimic p53’s action or identify other vulnerabilities that make these cells cancerous,” the author says. “These findings suggest that if p53’s job is to eliminate this plastic, injury repair state, then strategies to directly target this state and its protective niche could represent an innovative way to help the significant percentage of patients whose cancers have lost p53.”
https://www.cell.com/cell/fulltext/S0092-8674(26)00333-8
https://sciencemission.com/Oncogenic-and-tumor-suppressive-forces