
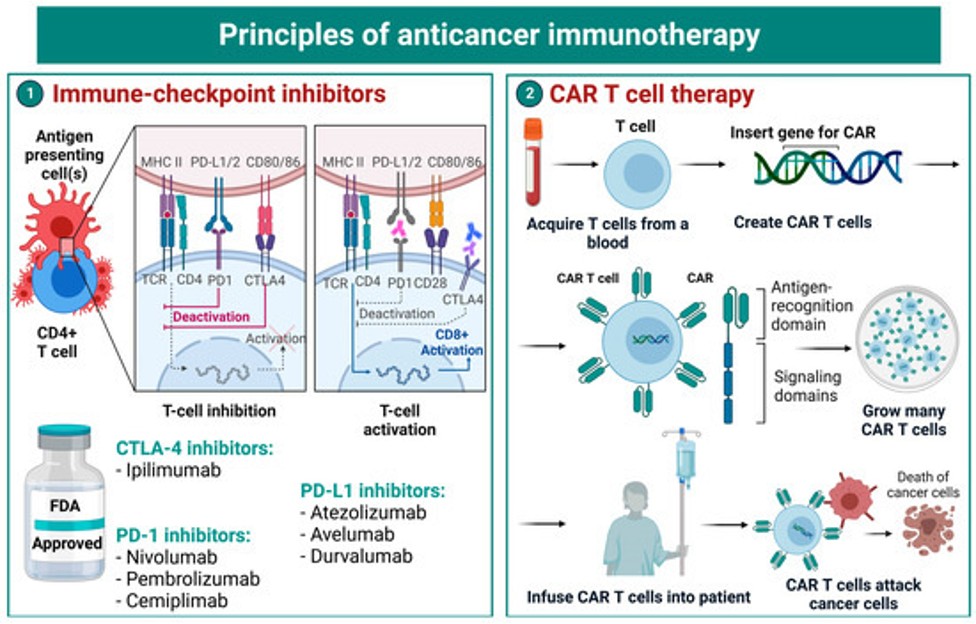

Cancer gene signatures in biopsy images using AI
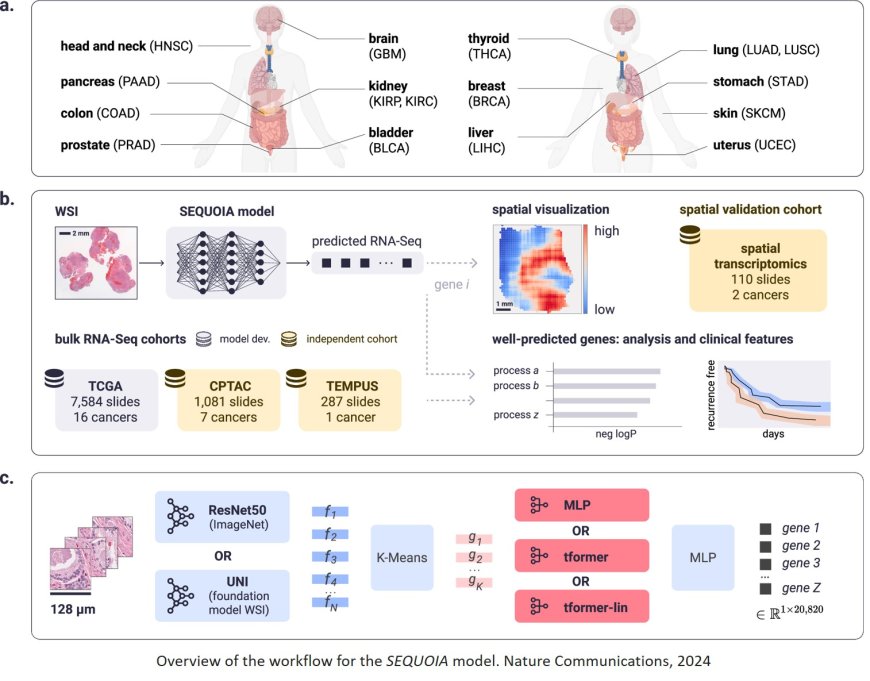
To determine the type and severity of a cancer, pathologists typically analyze thin slices of a tumor biopsy under a microscope. But to figure out what genomic changes are driving the tumor’s growth — information that can guide how it is treated — scientists must perform genetic sequencing of the RNA isolated from the tumor, a process that can take weeks and costs thousands of dollars.
Now, the researchers have developed an artificial intelligence-powered computational program that can predict the activity of thousands of genes within tumor cells based only on standard microscopy images of the biopsy. The tool, described in Nature Communications was created using data from more than 7,000 diverse tumor samples. The team showed that it could use routinely collected biopsy images to predict genetic variations in breast cancers and to predict patient outcomes.
“This kind of software could be used to quickly identify gene signatures in patients’ tumors, speeding up clinical decision-making and saving the health care system thousands of dollars,” said the senior author of the paper.
Clinicians have increasingly guided the selection of which cancer treatments — including chemotherapies, immunotherapies and hormone-based therapies — to recommend to their patients based on not only which organ a patient’s cancer affects, but which genes a tumor is using to fuel its growth and spread. Turning on or off certain genes could make a tumor more aggressive, more likely to metastasize, or more or less likely to respond to certain drugs.
However, accessing this information often requires costly and time-consuming genomic sequencing.
The researchers knew that the gene activity within individual cells can alter the appearance of those cells in ways that are often imperceptible to a human eye. They turned to artificial intelligence to find these patterns.
The researchers began with 7,584 cancer biopsies from 16 different of cancer types. Each biopsy had been sliced into thin sections and prepared using a method known as hematoxylin and eosin staining, which is standard for visualizing the overall appearance of cancer cells. Information on the cancers’ transcriptomes — which genes the cells are actively using — was also available.
After the researchers integrated their new cancer biopsies as well as other datasets, including transcriptomic data and images from thousands of healthy cells, the AI program — which they named SEQUOIA (slide-based expression quantification using linearized attention) — was able to predict the expression patterns of more than 15,000 different genes from the stained images. For some cancer types, the AI-predicted gene activity had a more than 80% correlation with the real gene activity data. In general, the more samples of any given cancer type that were included in the initial data, the better the model performed on that cancer type.
“It took a number of iterations of the model for it to get to the point where we were happy with the performance,” the author said. “But ultimately for some tumor types, it got to a level that it can be useful in the clinic.”
The author pointed out that doctors are often not looking at genes one at a time to make clinical decisions, but at gene signatures that include hundreds of different genes. For instance, many cancer cells activate the same groups of hundreds of genes related to inflammation, or hundreds of genes related to cell growth. Compared with its performance at predicting individual gene expression, SEQUOIA was even more accurate at predicting whether such large genomic programs were activated.
To make the data accessible and easy to interpret, the researchers programmed SEQUOIA to display the genetic findings as a visual map of the tumor biopsy, letting scientists and clinicians see how genetic variations might be distinct in different areas of a tumor.
To test the utility of SEQUOIA for clinical decision making, the researchers identified breast cancer genes that the model could accurately predict the expression of and that are already used in commercial breast cancer genomic tests. (The Food and Drug Administration-approved MammaPrint test, for instance, analyzes the levels of 70 breast-cancer-related genes to provide patients with a score of the risk their cancer is likely to recur.)
“Breast cancer has a number of very well-studied gene signatures that have been shown over the past decade to be highly correlated with treatment responses and patient outcomes,” the author said. “This made it an ideal test case for our model.”
SEQUOIA, the team showed, could provide the same type of genomic risk score as MammaPrint using only stained images of tumor biopsies. The results were repeated on multiple different groups of breast cancer patients. In each case, patients identified as high risk by SEQUOIA had worse outcomes, with higher rates of cancer recurrence and a shorter time before their cancer recurred.
The AI model can’t yet be used in a clinical setting — it needs to be tested in clinical trials and be approved by the FDA before it’s used in guiding treatment decisions — but the team is improving the algorithm and studying its potential applications. In the future, the said, SEQUOIA could reduce the need for expensive gene expression tests.
“We’ve shown how useful this could be for breast cancer, and we can now use it for all cancers and look at any gene signature that is out there,” the author said. “It’s a whole new source of data that we didn’t have before.”