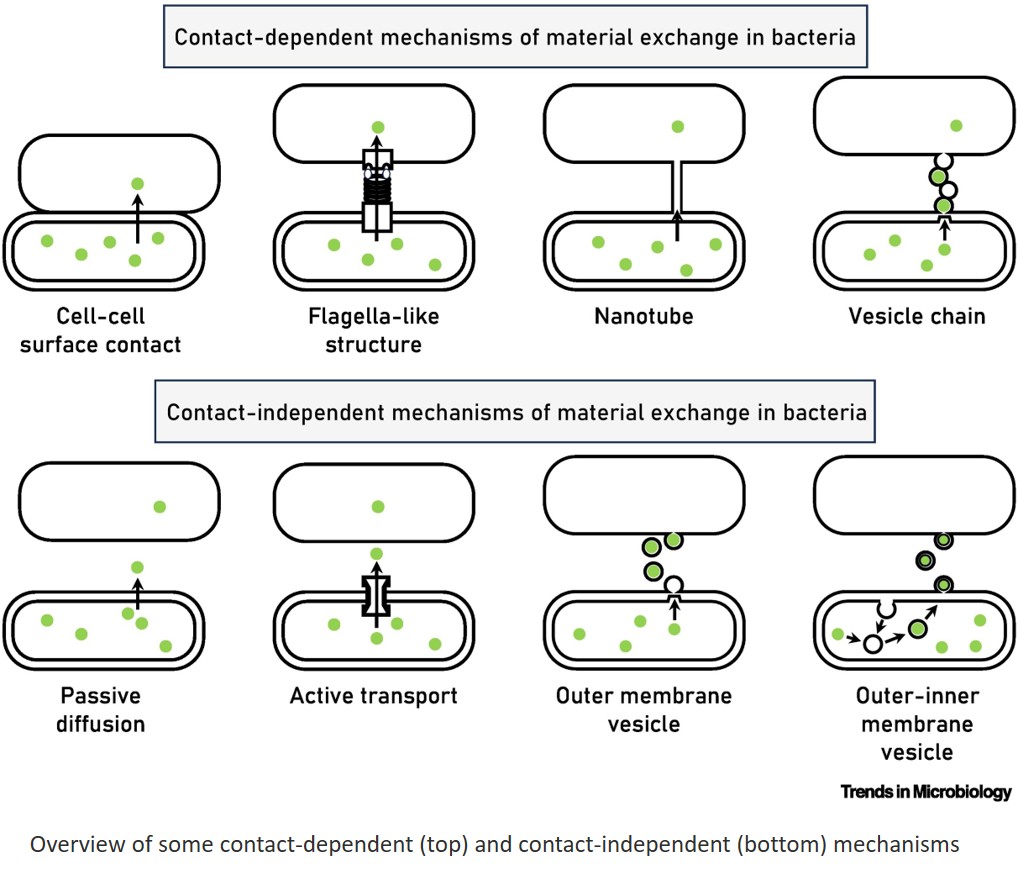
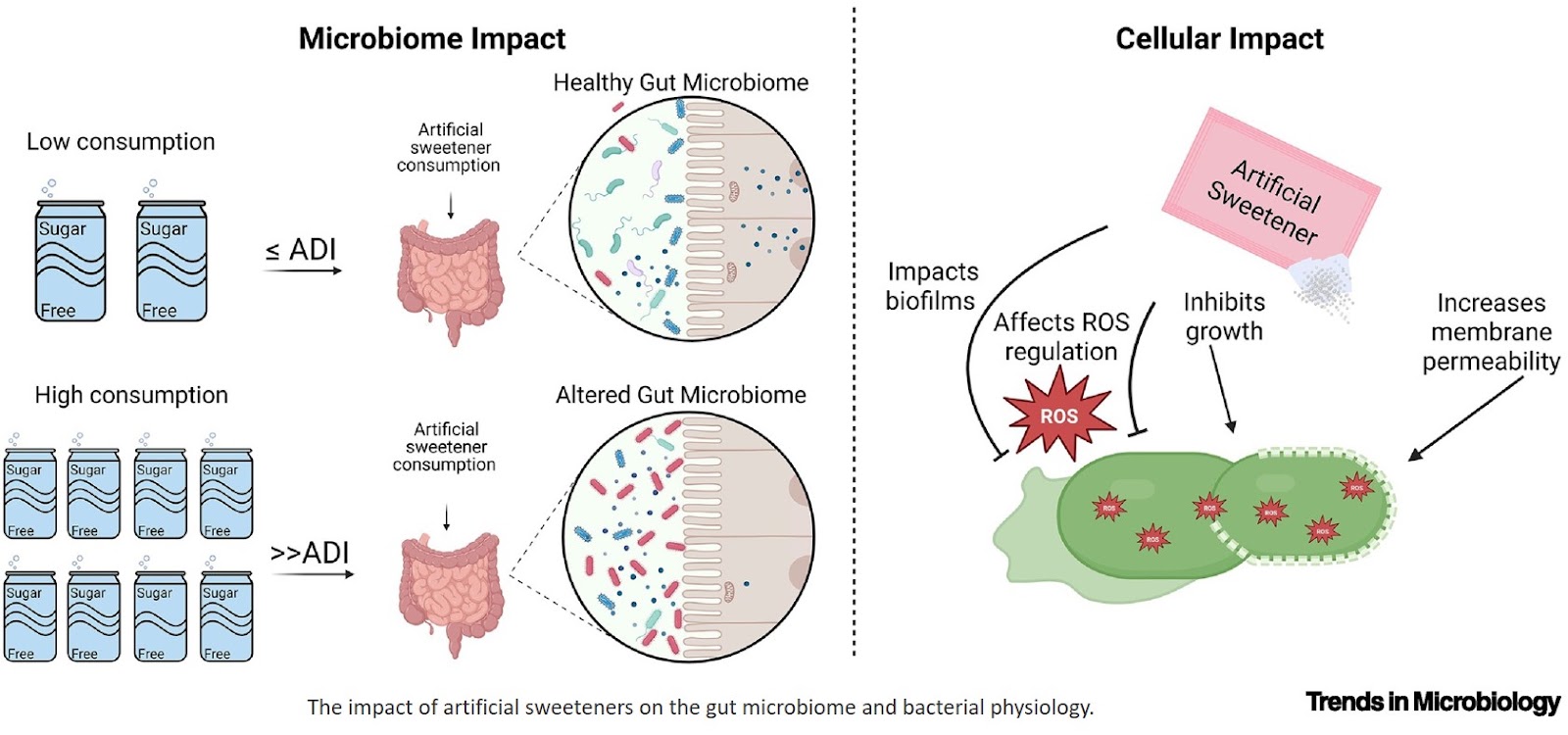
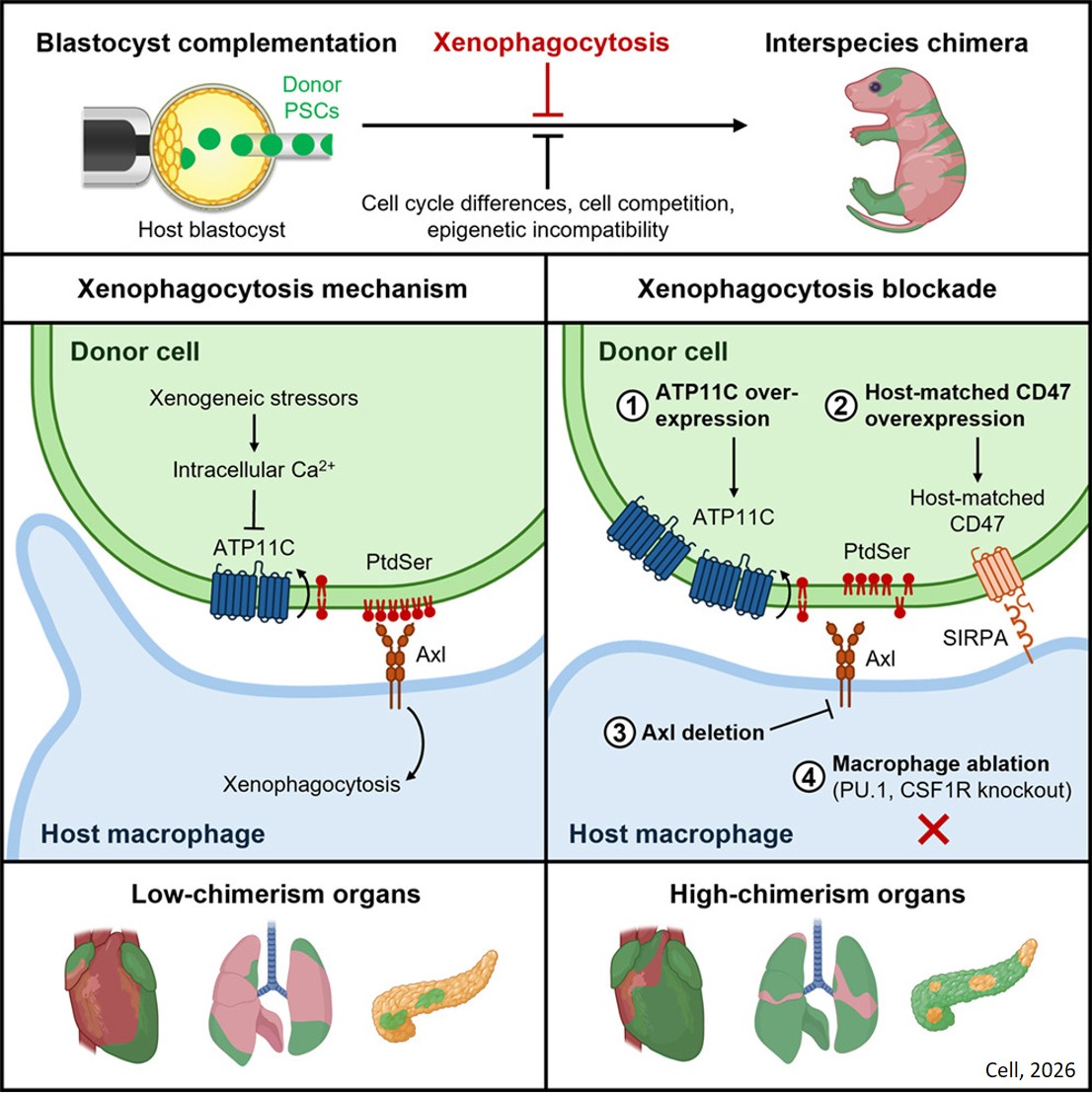
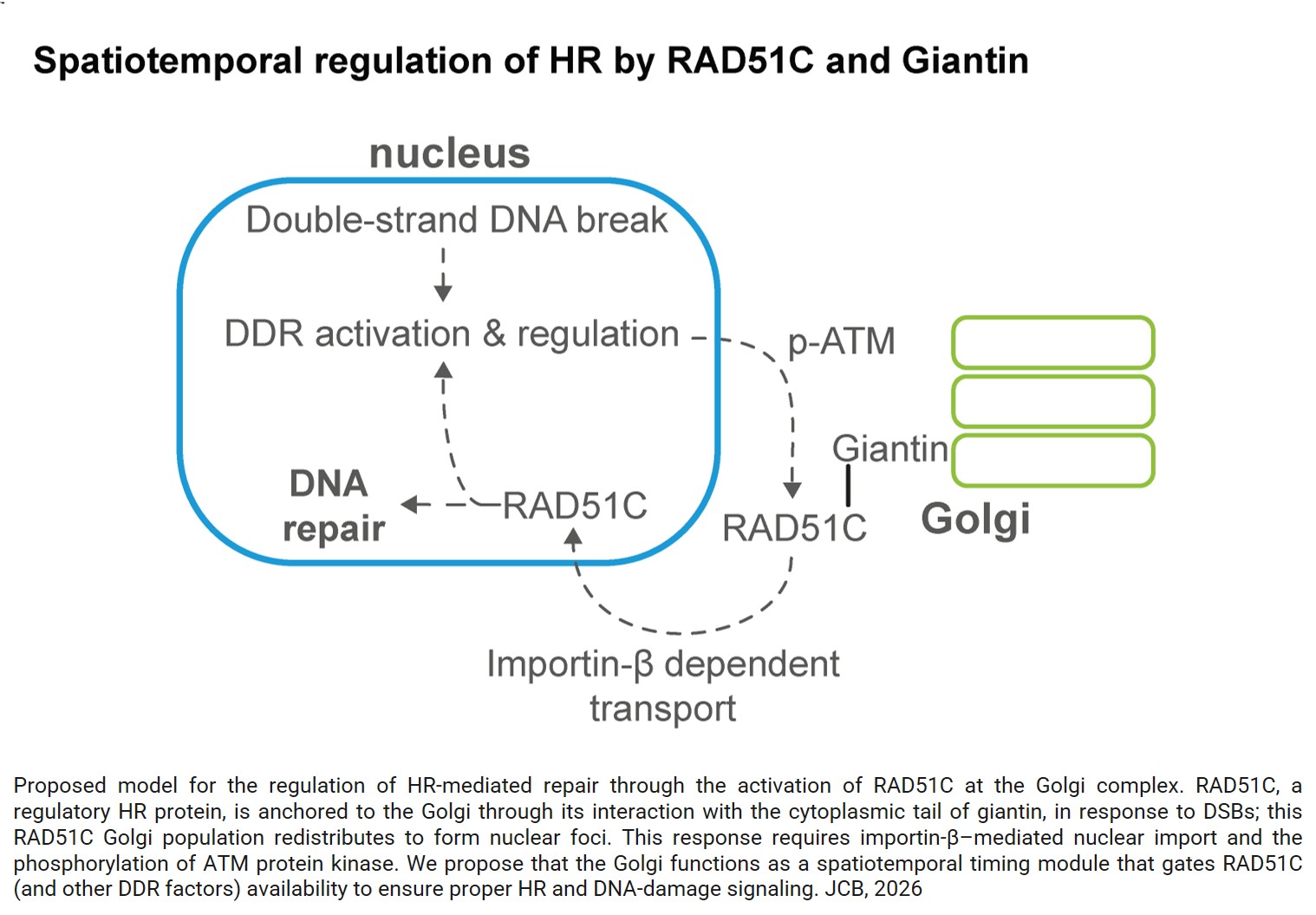


A gene regulatory protein identified as a novel dependency in cancers
As many as one in four cancers are driven by mutations in the SWI/SNF chromatin-remodeling complex, which controls access to DNA. A new study recently identified the gene-regulatory protein PHIP as a critical vulnerability in cancers driven by broad SWI/SNF inactivation. The work revealed PHIP as a potential therapeutic target for cancers, including rhabdoid tumors in children, and other hard-to-treat malignancies. The results were published in Nature Communications.
Cancers with SWI/SNF mutations are hard to target because parts of the complex that serve as tumor suppressors, such as SMARCB1, are lost. The challenge of having a missing target has pushed researchers to look for alternative proteins and pathways that cancer cells become dependent on when SWI/SNF is disrupted.
“We’re studying these cancers to understand the fundamental mechanisms that control the formation of these malignancies,” said corresponding author of the study. “When mutations disrupt SWI/SNF, we want to know what downstream processes drive cancer — and which of them can be targeted.”
“We’ve identified both a new target and a new mechanism of chromatin regulation that becomes critical when SWI/SNF is broadly inactivated,” added first-author of the study.
Rhabdoid tumors, fast-growing cancers affecting infants and young children, typically form in the kidneys, soft tissues, or central nervous system, where they are known as atypical teratoid rhabdoid tumor, or AT/RT. In 95% of cases, rhabdoid tumors are driven by loss of the SWI/SNF subunit SMARCB1. With few other mutations, these cancers offer a clear model for studying how disruption of SWI/SNF causes cancer and for identifying related vulnerabilities.
“We use rhabdoid tumors because they have a mutation in this SWI/SNF complex but an otherwise pristine genome,” said the author. “Adult cancers with SWI/SNF mutations have many other mutations that can make it hard to figure out what’s important. We’re learning that the vulnerabilities are proteins and processes that work closely with the SWI/SNF complex, either in cooperation or in opposition to it.”
To look for potential dependencies, the researchers used data from more than 1,000 cancer cell lines in the Cancer Dependency Map, a community portal developed through the Pediatric Cancer Dependencies Accelerator. This included 15 SMARCB1‑deficient rhabdoid tumor cell lines that the researchers had contributed. From this work, PHIP emerged as a top genetic dependency.
“It was not immediately clear why PHIP becomes essential when SMARCB1 is lost, but we learned that it cooperates with SWI/SNF to activate transcription,” said the author.
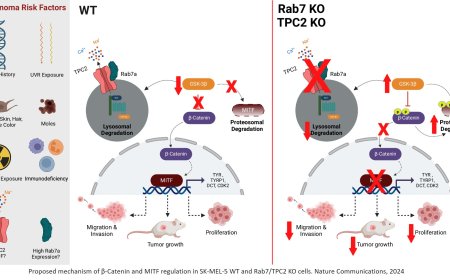
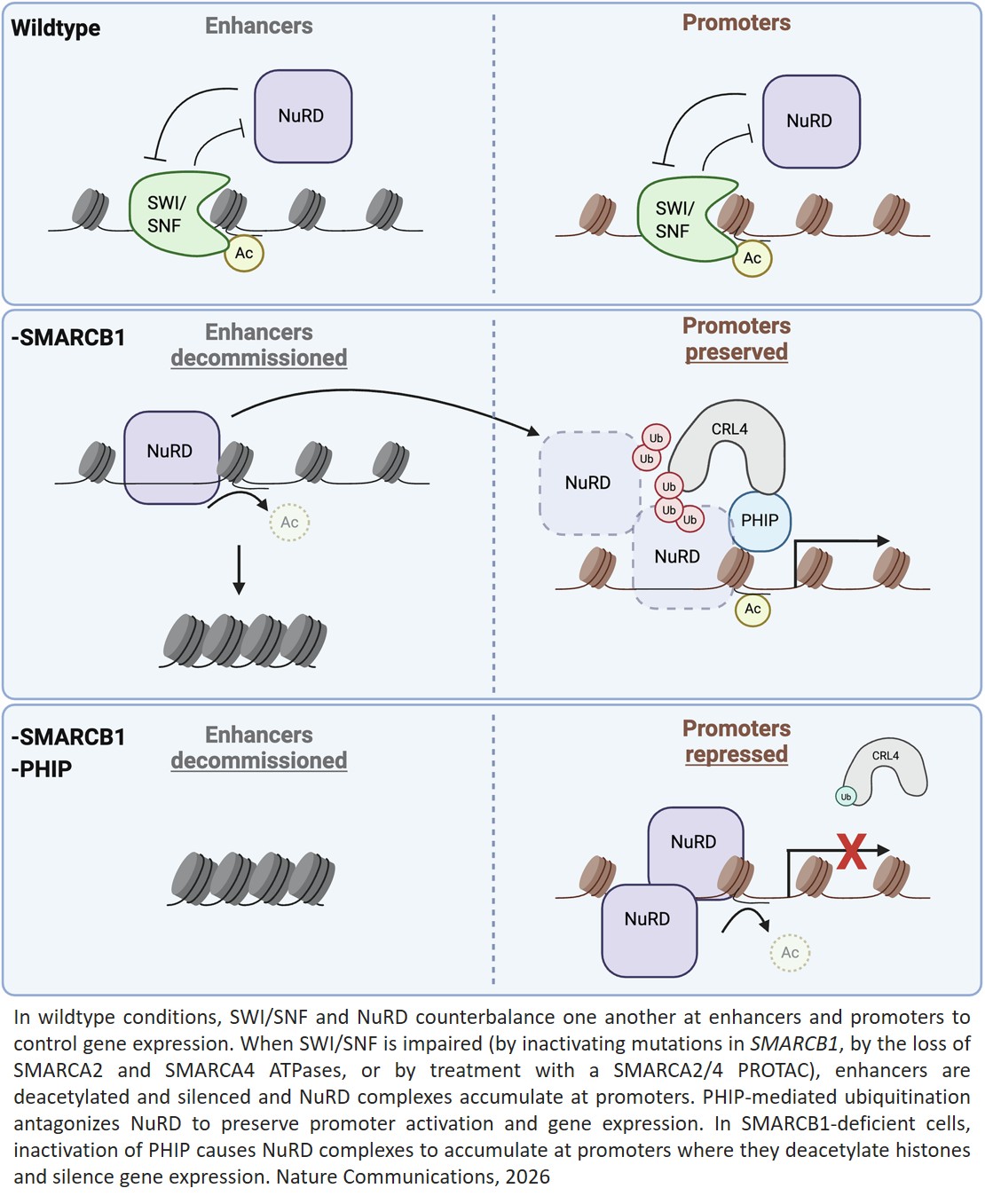
Using cell line and patient-derived xenograft and organoid models, the researchers uncovered how PHIP supports the growth of SWI/SNF-mutant cancers. SWI/SNF normally helps turn on genes, while an opposing complex, NuRD, silences them. PHIP cooperates with SWI/SNF to enable proper gene activation by suppressing NuRD.
“When the right genes are ready to go, you need SWI/SNF to turn on and NuRD to turn off,” said the author. “SWI/SNF overcomes NuRD repression so genes can be expressed. In highly proliferative progenitor cells, failure of this process can lead to cancer.”
“SWI/SNF and NuRD sit at the same genomic locations, but have opposite effects,” added the author. “When SWI/SNF is absent, PHIP becomes essential to hold NuRD’s silencing activity in check.”
There are no existing drugs targeting PHIP, but chemical inhibitors have been identified, and the findings provide a strong rationale for their development. “PHIP is overexpressed and associated with poor prognosis in several cancers,” said the author. “We know the balance between SWI/SNF and NuRD is crucial in development and disease, so targeting PHIP could offer a valuable therapeutic approach.”